|
- (Requires access to linux server with tandem programme installed)
- Login to iridis5_a.soton.ac.uk
- Download this zip file to your directory on iridis5_a.soton.ac.uk
- You'll see several xml files in the bin directory; for the purposes of this tutorial you do not need to edit these
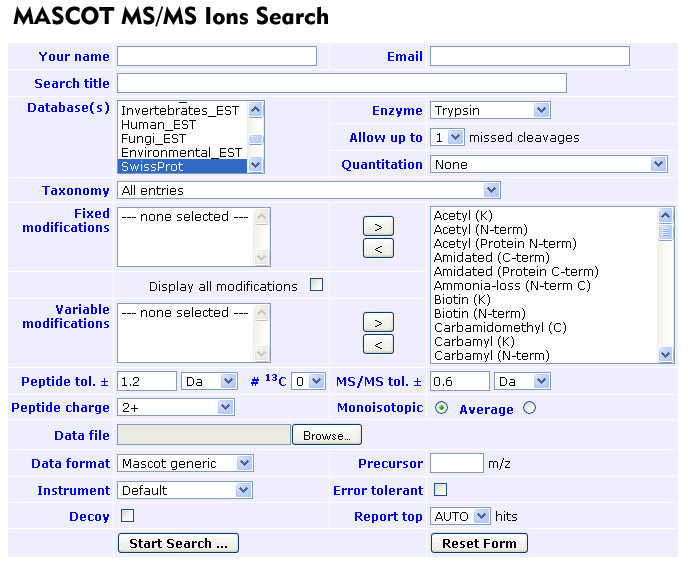
- Take a look at the default_input.xml file (this is where the parameters for ther tandem programme are set)
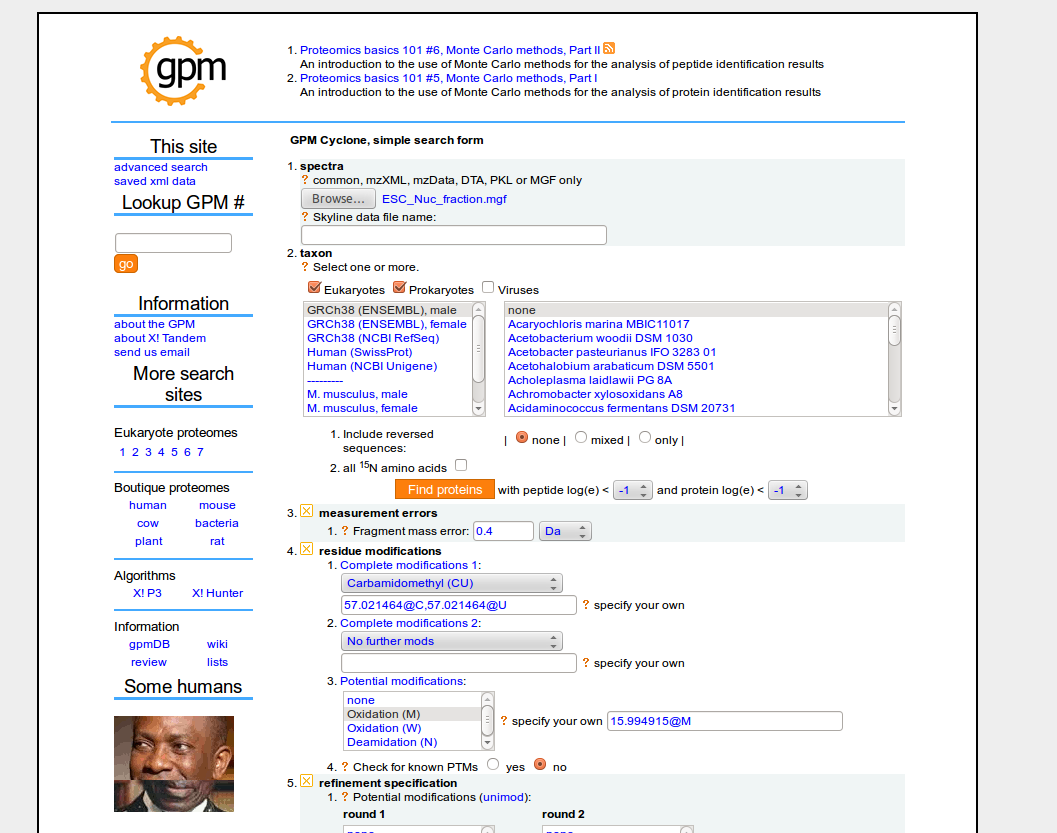
- To search your mass-spectra data:
- The output*xml files contain lots of information about the peptides and proteins that have been identified
- Here's some code to extract the proteins identified and their expect values (the statistical significance of the match):
awk 'BEGIN{OFS=","} /^<protein/ {print $5,$2}' output.*.xml | sed 's/\(expect=\|label=\|\"\| \)//g;' | sort -t , -k 2,2n
- The expect values are actually log(expect) - so the large more negative numbers indicate a more significant p-value
|